contigimage - create contig images based on .ace file
contigimage [ace_file]
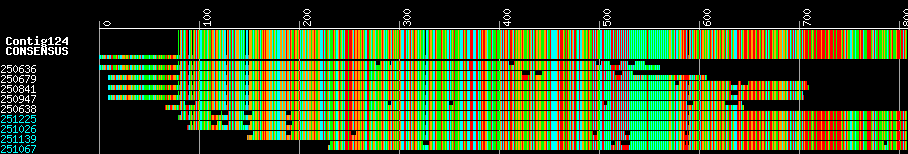
During DNA sequence processing, sequence fragments are aligned to create longer fragments based on overlap. The overlapping sequences are known as a contig. The contig is assembled from reads into a consensus sequence. The program phrap does the assembly. Contigimage parses the .screen.ace.1 output file from phrap and generates contig images, one per contig, storing them in PNG format in files according to contig name. The consensus sequence is on top, each sequence in the assembly follows below, where the name of the sequence is on the left (in cyan if it was reverse complemented), the base pair position is on the X axis, the quality is the height of the lane, and the color scheme is like this.
[A = green, C = cyan, G = orange, T=red]
[N = gray, X = darkgray, * = darkgray]
It also creates a thumbnail image if the number of reads in the contig is greater than four. The width of the thumbnail image is one quarter that of the normal image, the height is one pixel per read in the contig.
Example of contig assembly image
Contigimage is designed to work within the bioPROC tree structre. That is, it knows where to find the .ace file within bioPROC. Specifically it means that contigimage must be in a contig directory, and that it will create an image subdirectory named images for the contig images and thumbnails it creates.
[ace_file]
By default, contigimage will use the ace file in the contig set directory. Thanks to phrap, this file name ends in ``fasta.screen.ace.1''. The user can override the default by specifying an ace file on the command line.
Summarized format of the ace file.
For detailed info, see the consed(1) man page, about page 37
AS <number of contigs> <total number of reads in ace file> CO <contig name> <#bases> <#reads> <#base segments> <U or C> lines of sequence data
BQ lines of sequence quality data
AF <read name> <C or U> <padded start consensus position>
BS <start position> <end position> <read name>
RD <read name> <# of padded bases> <# of read info items> <# of read tags>
QA <start> <end> <align start> <align end>
DS CHROMAT_FILE: <name > PHD_FILE: <name > TIME: <date/time phd file>
WR { <tag type> <program > <YYMMDD:HHMMSS> }
RT{ <tag type> <program > < start> <end> <YYMMDD:HHMMSS> }
CT{ <contig name> <tag type> <program> <start> <end> <YYMMDD> (info) }
WA{ <tag type> <program> <YYMMDD:HHMMSS> 1 or more lines of data }
CO segments mark the beginning of a contig description. There will be one BQ segment associated with each CO, and one or more AF, and RD segments associated with each CO. Contigimage only looks at CO, BQ, AF, and RD segments.
biodata2crawler(1), biodata2recurse(1), phrap(1), consed(1), gelimage(1)